 Professor
Professor
Research Areas
Structural analysis of transcription
Research Interests
 and EC(B) structures.") Fig. 1.The ttRNAP holo-enzyme(A) and EC(B) structures.Protein crystallography provides the most detailed and compelling account of the structure and function of biological macromolecules and leads to a better understanding of all cellular processes. Building on the experience we have accumulated during previous crystallographic studies covering a wide range of proteins, protein-ligand and protein-nucleic acid complexes from various biological pathways, since 2002 my lab focused on structural analysis of transcription, using crystallography as a tool and RNA polymerase (RNAP) as a major target. Crystallographic studies of transcription are particularly challenging, as most of the proteins from this pathway appear to be very flexible and unstable, thus being refractory to crystallizations and structure determination. Together with the ribosome, multi-subunit RNAPs are among the biggest asymmetric macromolecules studied by X-ray diffraction so far and are therefore at the cutting edge of modern protein crystallography.
Fig. 1.The ttRNAP holo-enzyme(A) and EC(B) structures.Protein crystallography provides the most detailed and compelling account of the structure and function of biological macromolecules and leads to a better understanding of all cellular processes. Building on the experience we have accumulated during previous crystallographic studies covering a wide range of proteins, protein-ligand and protein-nucleic acid complexes from various biological pathways, since 2002 my lab focused on structural analysis of transcription, using crystallography as a tool and RNA polymerase (RNAP) as a major target. Crystallographic studies of transcription are particularly challenging, as most of the proteins from this pathway appear to be very flexible and unstable, thus being refractory to crystallizations and structure determination. Together with the ribosome, multi-subunit RNAPs are among the biggest asymmetric macromolecules studied by X-ray diffraction so far and are therefore at the cutting edge of modern protein crystallography.
The structures of the single-subunit T7 RNAP elongation complexes (ECs) revealed for that transition from initiation to elongation phase in this system is accompanied by the unprecedented structural alterations of the enzyme, while the NTP-substrate loading is mediated by the two, preinsertion (open, inactive) and insertion (closed, active) states. The 2.6Å resolution structure of the multi-subunit bacterial RNAP holo-enzyme (Fig. 1A) provided deep insights into the crucial mechanisms of initiation. The structures of the bacterial ECs (Fig. 1B) provided the high resolution (2.5Å) data for the multi-subunit enzymes and revealed many important fundamental mechanisms: the two step mode of substrate loading, active site closure via the folding of the trigger loop (TL), bubble formation, translocation and catalytic reaction.
Bacterial RNAP is an attractive target for the design of antibiotics. First, despite the overall homology in structure and function of bacterial and eukaryotic RNAPs, each exhibits many distinct features, in particular, in the regulation of transcription. Second, consisting of over 3,000 amino acids, bacterial RNAP comprises a huge solvent-exposed surface that contains numerous cavities and channels, many of which are used for binding nucleic acids and/or transcription factors. Blocking off these cavities with a steric inhibitor may therefore result in disruption of transcription and eventually cell death. In order to utilize structure-based drug design of a novel antibiotics targeting RNAP one should understand the molecular mechanism of their action for which determination of the atomic structures of RNAP in complexes with the promising inhibitors is of central importance. The structures of RNAP complexed with small inhibitors/antibiotics not only provided a short-cut to rational drug design, but also captured snap-shots of important dynamic intermediates. The structures of the Dks Aprotein and RNAP complexed with "magic spot" (ppGpp) provided insights in the DksA/ppGpp synergism during stringent control. The RNAP complexes with clinically important rifamycins revealed antibiotic-induced allosteric remodeling of the RNAP active site. The streptolydigin-bound intermediates provided the first evidence for the substrate preinsertion state in the mulit-subunit RNAPs and for the key regulatory role of the TL. Finally, the RNAP complex with myxopyronin revealed a crucial, active role of the RNAP core-enzyme during open complex formation and a local conformational switch in RNAP as a checkpoint in this process. These works shed considerable light on the mechanistic aspects of the transcription regulation by these compounds thereby allowing intellectual improvement of their inhibitory properties. The high-resolution 3D structures of the transcription complexes that we have so far determined revealed mechanistic details that are indispensable for bridging the gap in our understanding of enzymatic features in many biological systems. However, obtaining only a single or few structure even of the key functional module(s) is only half-the-battle. Function of transcription machinery is accompanied by structural alterations and formation of many transient complexes that can be elucidated only through comparative structural analyses of a series of functional intermediates. Given the huge number of such intermediates formed, for example, by RNAP studies of functionally significant transcription complexes take on a massive scale similar to that of structural genomics projects. Our major ultimate goal is to eventually assemble a comprehensive functional “movie” of the dynamic and tightly regulated transcription process from serial high-resolution crystallographic snapshots. To achieve this goal we plan to expand our efforts towards building a high-throughput platform for structural and functional studies of the multiple transcription intermediates. The major challenge in establishing this platform is that the three key steps of crystallographic studies (expression, purification, crystallization) are performed by the tricky, unpredictable trial-and-error algorithms. Our goal therefore, is to develop a set of systematic, largely target-independent and ideally, high-throughput approaches to expression, purification and crystallizationof the complex biological molecules.
Protein purification is an essential primary step in numerous biological studies. Obtaining the protein samples with High yield, High purity and High activity (HHH-purification) is compulsory for determination of high-resolution 3D structures. An extra-High purity is especially critical for successful crystallization since even subtle (3-10%) contaminations may hinder crystal formation or affect diffraction quality of the crystals. However, protein expression and purification is a target-specific, multi-step and often poorly reproducible process, which is particularly challenging for multi-subunit proteins. For example, though many groups worldwide studied bacterial RNAPs from different organisms for decades, no universal and efficient multi-subunit expression vector or HHH-purification protocol were developed based on the available expression/purification systems.
To advance expression and purification of the transcription complexes and other challenging biological systems we have constructed and efficient multi-subunit expression vector and developed a novel chromatographic technology based on the ultra-high affinity complex between the Colicin E7 DNAse (CE7) and its inhibitor, Immunity protein 7 (Im7). To avoid toxicity of a natural DNAse domain, we mutated CE7 to create an inactive CL7-tag (~16kDa), which retained the full binding affinity to Im7 but was inactivated as a DNAse. To achieve high capacity, we developed a protocol for a large-scale production and highly specific immobilization of Im7 (~12 kDa) to a solid support. To challenge our system, we applied it to the complex targets from the major categories (membrane, toxic, multi-subunit DNA-binding proteins), which are most refractory to HHH-purification.
Membrane proteins: The HHH-purification of the membrane proteins is always challenging due to their unique hydrophobic nature and poor over-expression levels. To test the CL7/Im7 system, we have selected two MPs from prokaryotic (Yidc membrane integrase; ~32kDa) and eukaryotic (Calnexin, CNX, ~65 kDa) organisms, which also drastically differ in their configuration, size and function. 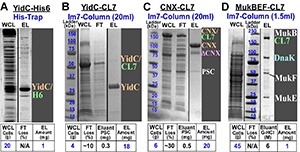 Figure 2Yidc was previously purified via a complex multi-step protocol to yield only ~1mg pure protein from ~15-20g cells in ~4 days. In particular, the first His-Trap step resulted in only ~60% purity (Fig. 2A). The full length CNX has never been over-expressed in E. coli or purified in the large (crystallographic) quantities. Using the CL7/Im7 purification system, the HHH-protein samples of Yidc and CNX were then obtained using a one-step Im7-run in an identical manner (Figs. 2B & 2C). The Im7-column demonstrated dramatic ~270 fold increase in productivity over the conventional techniques.
Figure 2Yidc was previously purified via a complex multi-step protocol to yield only ~1mg pure protein from ~15-20g cells in ~4 days. In particular, the first His-Trap step resulted in only ~60% purity (Fig. 2A). The full length CNX has never been over-expressed in E. coli or purified in the large (crystallographic) quantities. Using the CL7/Im7 purification system, the HHH-protein samples of Yidc and CNX were then obtained using a one-step Im7-run in an identical manner (Figs. 2B & 2C). The Im7-column demonstrated dramatic ~270 fold increase in productivity over the conventional techniques.
Toxic proteins: Many functionally significant proteins may be toxic for cells or possess some folding defects, when over-expressed. These proteins may be isolated only directly from the host cells. However, such purification is problematic since exceedingly low expression levels are often beyond sensitivity of the existing purification techniques. The bacterial multi-subunit MukBEF condensin complex (~526 kDa) compacts cellular DNA into nucleoids and chromosomes. Its over-expression is toxic to the cells, while expression from the chromosome is very low (Fig. 2D, WCL). We labeled the MukB gene with the C-terminal CL7-tag in its genomic locus. We loaded the lysate of ~45 g mutated cells on a small (1.5 ml) Im7-column and purified ~1mg of the high purity (~97%) complex in a single step (Fig. 2D). These results show that the CL7/Im7 system may achieve HHH-purification of the most complex, poorly characterized proteins expressed at the very low levels.
Multi-subunit RNAPs: So far, HHH-purification of RNAPs is a multi-step, specie-specific process. For example, the core enzymes of T. thermophilus and M. tuberculosis RNAPs are distinct in configuration and surface properties. Thus, these RNAPs represent essentially distinct targets for both expression and purification that is reflected in the largely different previously established purification procedures. We have designed a universal multi-subunit expression vector with enhanced expression levels of the individual subunits and the protease cleavable CL7-tag at the C-terminus of the largest subunit. 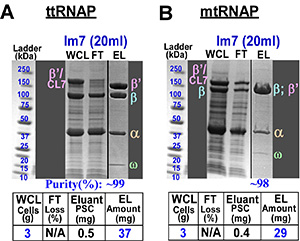 Figure 3As a result, we succeeded in an essentially identical one-step HHH-purification protocols for both enzymes using the CL7/Im7 approach to obtain a large amounts of a ~99% pure RNAP samples (Fig. 3). To conclude, our multi-subunit expression and Im7-purification systems universally provided a dramatic (10-500 fold) improvement over the conventional protocols for the two evolutionary distinct RNAPs.
Figure 3As a result, we succeeded in an essentially identical one-step HHH-purification protocols for both enzymes using the CL7/Im7 approach to obtain a large amounts of a ~99% pure RNAP samples (Fig. 3). To conclude, our multi-subunit expression and Im7-purification systems universally provided a dramatic (10-500 fold) improvement over the conventional protocols for the two evolutionary distinct RNAPs.
In Summary: The Im7-column allows for one-step HHH-purification of challenging proteins, including RNAPs. It has a high capacity, stringent affinity to only its cognate CL7-domain and tolerates high-salt during lysate loading. The latter is imperative for NA-binding proteins, such as RNAPs. The CL7/Im7 approach forms a solid basis for a proposed high-throughput platform. The CL7/Im7 system is simple, reusable and is also applicable to pull-down and kinetic activity/binding assays.
Education
Graduate School
Ph.D., Chemistry, Institute of Molecular Biology of the Academy of Sciences of USSR (Moscow)
Postdoctoral Fellowship
Institute of Molecular Biology of the Academy of Sciences of USSR (Moscow)
Protein Engineering Institute (Osaka, Japan)
Contact
Office
Kaul Human Genetics Building
Building Room 440D
720 20th Street South
Birmingham, AL 35233
Phone
(205) 807-6096
Email
dmitry@uab.edu