Stacey Harbour, Ph.D., Scientist I, Anatomic Pathology, in the lab of Casey Weaver, M.D., Professor, Anatomic Pathology, published an article on T cell differentiation in the July issue of Science Immunology, titled, "TH17 cells require ongoing classic IL-6 receptor signaling to retain transcriptional and functional identity."
Research conducted by the group showed that the molecule IL-6, a common target of various disease therapies, was indispensable for maintaining the inflammatory effects of T helper 17 cells that are implicated in diseases like IBD and psoriasis.
The article is published with co-authors Robin Hatton, Ph.D., Associate Professor, Anatomic Pathology; Daniel DiToro, Ph.D., Graduate Student, Anatomic Pathology; Steven Witte, Ph.D., Consultant, Anatomic Pathology; Carlene Zindl, Scientist I, Anatomic Pathology; Min Gao, Ph.D., Assistant Professor, Medicine; Trenton Schoeb, Ph.D., Professor, Genetics.
From the article:
"The pathogenic role of the TH17 subset of CD4+ T cells in multiple immune-mediated diseases has prompted close scrutiny of the cytokine signals that promote differentiation and maintenance of TH17 cells. IL-6 and TGF-β are key cytokines for TH17 commitment by naïve T cells whereas IL-23 supports maintenance of a TH17 identity. Harbour et al. investigated whether persistent IL-6 signaling is also needed to sustain TH17 cell functions. IL-6Rα–deficient mouse T cells could not maintain a TH17 phenotype and were attenuated in their ability to elicit colitis in an in vivo cell transfer model. These studies provide deeper insights into the set of signals required for TH17 cell maintenance and suggest additional molecular targets for pharmacological interventions aimed at antagonizing pathogenic TH17 immunity.
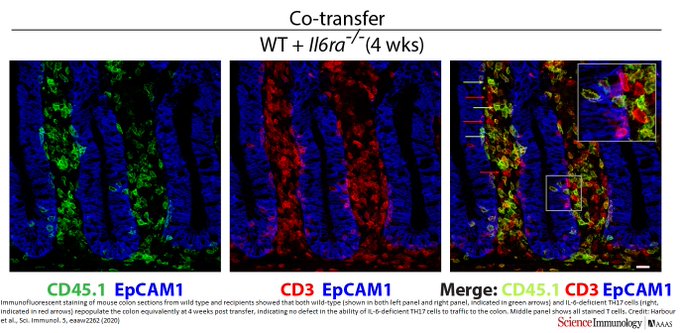
Acting in concert with TGF-β, interleukin-6 (IL-6) signaling induces T helper 17 (TH17) cell development by programming TH17-related genes via signal transducers and activators of transcription 3 (STAT3). A role for IL-6 signaling beyond the inductive phase of TH17 cell development has not been defined because IL-23 signaling downstream of TH17 cell induction also activates STAT3 and is thought responsible for TH17 cell maintenance. Here, we find that IL-6 signaling is required for both induction and maintenance of mouse TH17 cells; IL-6Rα–deficient TH17 cells rapidly lost their TH17 phenotype and did not cause disease in two models of colitis. Cotransfer of wild-type TH17 cells with IL-6Rα–deficient TH17 cells induced colitis but was unable to rescue phenotype loss of the latter. High IL-6 expression in the colon promoted classic, or cis, rather than transreceptor signaling that was required for maintenance of TH17 cells. Thus, ongoing classic IL-6 signaling underpins the TH17 program and is required for TH17 cell maintenance and function."
Look for a full news release to come.